Welcome to Dokhlab's Research Portal
The Dokhlab at Penn State College of Medicine focuses on computational approaches to understand complex biological systems. Our research spans molecular modeling, drug discovery, protein engineering, and RNA dynamics.
About us
About us
About us
Research Focus
Research Focus
Our Research Focus
Our laboratory develops and applies computational methods to investigate:
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Molecular dynamics and enhanced sampling techniques
Protein folding and misfolding mechanisms
Structure-based drug design
Protein-protein and protein-ligand interactions
RNA structure prediction and analysis
Current Projects
We actively collaborate with experimental laboratories to validate computational predictions and drive new discoveries in molecular biology and biophysics.
Searching for methyllysine-binding aromatic cages
Searching for methyllysine-binding aromatic cages
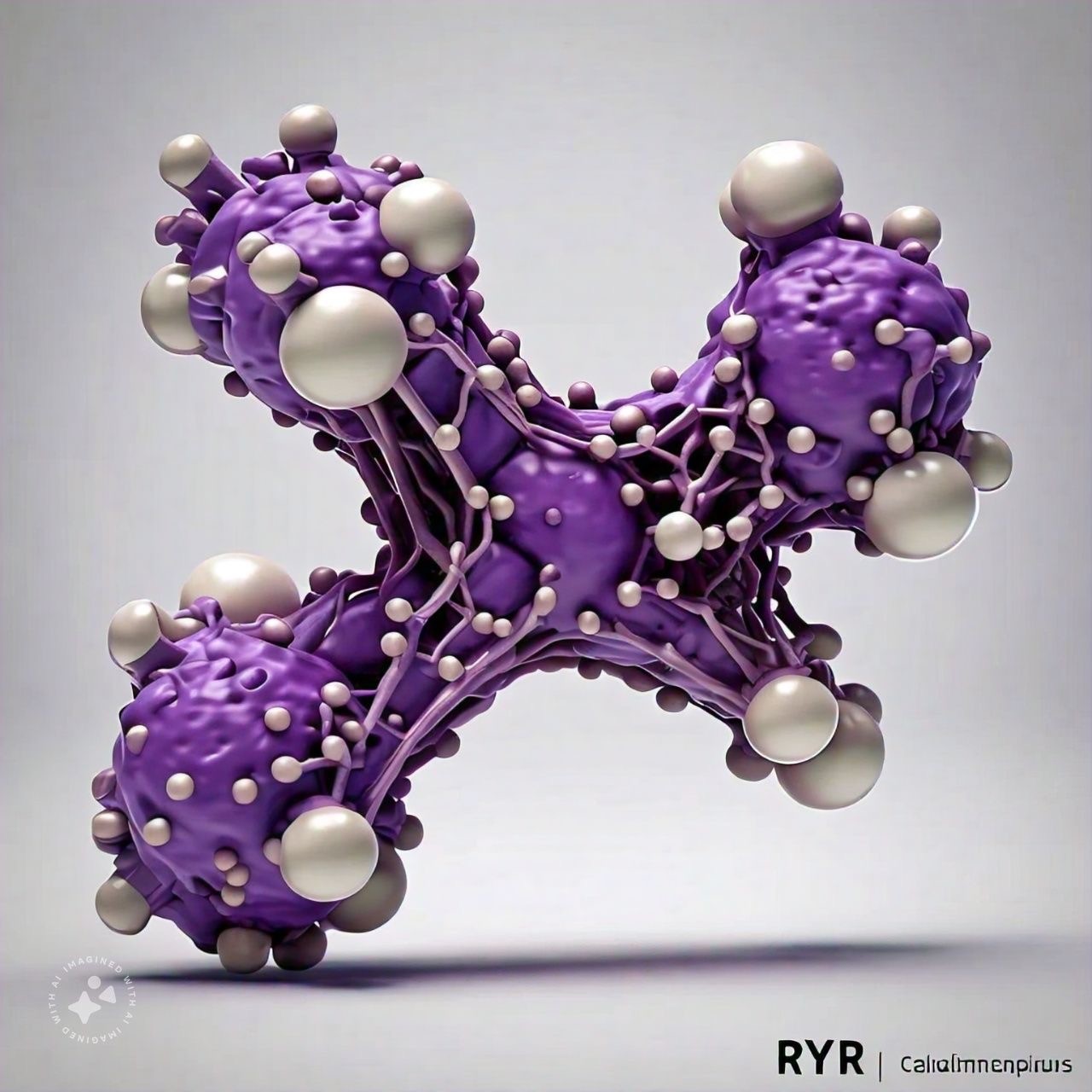
Ryanodine receptor 1 (RyR1) is an intracellular calcium ion (Ca2+) release channel required for skeletal muscle contraction. Although cryo-electron microscopy identified binding sites of three coactivators Ca2+, ATP and caffeine (CFF), the mechanism of co-regulation and synergy of these activators is unknown. Here, we report allosteric connections among the three ligand binding sites and pore region in (i) Ca2+ bound-closed, (ii) ATP/CFF bound- closed, (iii) Ca2+/ATP/CFF bound-closed, and (iv) Ca2+/ATP/CFF bound-open RyR1 states. We identified two dominant interactions that mediate interactions between the Ca2+ binding site and pore region in Ca2+ bound-closed state, which partially overlapped with the pore communications in ATP/CFF bound-closed RyR1 state. In Ca2+/ATP/CFF bound-closed and -open RyR1 states, co-regulatory interactions were analogous to communications in the Ca2+ bound-closed and ATP/CFF bound- closed states.
Ryanodine receptor 1 (RyR1) is an intracellular calcium ion (Ca2+) release channel required for skeletal muscle contraction. Although cryo-electron microscopy identified binding sites of three coactivators Ca2+, ATP and caffeine (CFF), the mechanism of co-regulation and synergy of these activators is unknown. Here, we report allosteric connections among the three ligand binding sites and pore region in (i) Ca2+ bound-closed, (ii) ATP/CFF bound- closed, (iii) Ca2+/ATP/CFF bound-closed, and (iv) Ca2+/ATP/CFF bound-open RyR1 states. We identified two dominant interactions that mediate interactions between the Ca2+ binding site and pore region in Ca2+ bound-closed state, which partially overlapped with the pore communications in ATP/CFF bound-closed RyR1 state. In Ca2+/ATP/CFF bound-closed and -open RyR1 states, co-regulatory interactions were analogous to communications in the Ca2+ bound-closed and ATP/CFF bound- closed states.
More Information
More Information
More Information
More Information

Inorganic meets organic programming
Inorganic meets organic programming
Virtual screening is a key enabler of computational drug discovery and requires accurate and efficient structure-based molecular docking. In this work, we develop algorithms and software building blocks for molecular docking that can take advantage of graphics processing units (GPUs). Specifically, we focus on MedusaDock, a flexible protein-small molecule docking approach and platform. We accelerate the performance of the coarse docking phase of MedusaDock, as this step constitutes nearly 70% of total running time in typical use-cases. We perform a comprehensive evaluation of the quality and performance with single-GPU and multi-GPU acceleration using a data set of 3875 protein-ligand complexes. The algorithmic ideas, data structure design choices, and performance optimization techniques shed light on GPU acceleration of other structure-based molecular docking software tools.
Virtual screening is a key enabler of computational drug discovery and requires accurate and efficient structure-based molecular docking. In this work, we develop algorithms and software building blocks for molecular docking that can take advantage of graphics processing units (GPUs). Specifically, we focus on MedusaDock, a flexible protein-small molecule docking approach and platform. We accelerate the performance of the coarse docking phase of MedusaDock, as this step constitutes nearly 70% of total running time in typical use-cases. We perform a comprehensive evaluation of the quality and performance with single-GPU and multi-GPU acceleration using a data set of 3875 protein-ligand complexes. The algorithmic ideas, data structure design choices, and performance optimization techniques shed light on GPU acceleration of other structure-based molecular docking software tools.
More Information
More Information
More Information
More Information
GPU-accelerated flexible molecular docking
GPU-accelerated flexible molecular docking
Virtual screening is a key enabler of computational drug discovery and requires accurate and efficient structure-based molecular docking. In this work, we develop algorithms and software building blocks for molecular docking that can take advantage of graphics processing units (GPUs). Specifically, we focus on MedusaDock, a flexible protein-small molecule docking approach and platform. We accelerate the performance of the coarse docking phase of MedusaDock, as this step constitutes nearly 70% of total running time in typical use-cases. We perform a comprehensive evaluation of the quality and performance with single-GPU and multi-GPU acceleration using a data set of 3875 protein-ligand complexes. The algorithmic ideas, data structure design choices, and performance optimization techniques shed light on GPU acceleration of other structure-based molecular docking software tools.
Virtual screening is a key enabler of computational drug discovery and requires accurate and efficient structure-based molecular docking. In this work, we develop algorithms and software building blocks for molecular docking that can take advantage of graphics processing units (GPUs). Specifically, we focus on MedusaDock, a flexible protein-small molecule docking approach and platform. We accelerate the performance of the coarse docking phase of MedusaDock, as this step constitutes nearly 70% of total running time in typical use-cases. We perform a comprehensive evaluation of the quality and performance with single-GPU and multi-GPU acceleration using a data set of 3875 protein-ligand complexes. The algorithmic ideas, data structure design choices, and performance optimization techniques shed light on GPU acceleration of other structure-based molecular docking software tools.
More Information
More Information
More Information
More Information

Methodologies
Our laboratory focuses on developing and applying computational methods to understand complex biological systems. Our primary research areas include:
Many biological processes occur at time scales not accessible to traditional simulation approaches.
Molecular Modeling and Simulations
Many biological processes occur at time scales not accessible to traditional simulation approaches.
Molecular Modeling and Simulations
Many biological processes occur at time scales not accessible to traditional simulation approaches.
Molecular Modeling and Simulations
Many biological processes occur at time scales not accessible to traditional simulation approaches.
Molecular Modeling and Simulations
Uncovering structures of molecular complexes via computational docking is at the heart of virtual drug screening and many structural modeling efforts.
Drug Discovery
Uncovering structures of molecular complexes via computational docking is at the heart of virtual drug screening and many structural modeling efforts.
Drug Discovery
Uncovering structures of molecular complexes via computational docking is at the heart of virtual drug screening and many structural modeling efforts.
Drug Discovery
Uncovering structures of molecular complexes via computational docking is at the heart of virtual drug screening and many structural modeling efforts.
Drug Discovery
Mutagenesis is a valuable exploratory tool in molecular biology and biotechnology.
Protein Design of Tools for Sensing and Controlling Proteins
Mutagenesis is a valuable exploratory tool in molecular biology and biotechnology.
Protein Design of Tools for Sensing and Controlling Proteins
Mutagenesis is a valuable exploratory tool in molecular biology and biotechnology.
Protein Design of Tools for Sensing and Controlling Proteins
Mutagenesis is a valuable exploratory tool in molecular biology and biotechnology.
Protein Design of Tools for Sensing and Controlling Proteins
RNA molecules are known for their central roles in gene expression, splicing, and translation.
RNA structure and dynamics
RNA molecules are known for their central roles in gene expression, splicing, and translation.
RNA structure and dynamics
RNA molecules are known for their central roles in gene expression, splicing, and translation.
RNA structure and dynamics
RNA molecules are known for their central roles in gene expression, splicing, and translation.
RNA structure and dynamics
Faculty Members
Discover how our platform has helped businesses create outstanding content effortlessly. Hear directly from our users about their success and satisfaction.

Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.

Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.

Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.

Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.

Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Dr Nikolay Dokholyan
Dr. Dokholyan's work spanned the area of statistical mechanics and its applications to biological macromolecules.
Jian Wang
They focus on specific research questions, conduct experiments, and contribute to publications and conference presentations.
Martin Dokholyan (High School Student)
Martin gains early exposure to scientific research by assisting with basic lab tasks and learning about computational biology.
Michelle Litvak
Michelle contributes by assisting with data collection and analysis, gaining hands-on experience in computational biology.
Brianna Hnath (Technician)
Brianna supports lab operations by maintaining equipment, managing supplies, and assisting in experimental procedures.
Our Labs Collaborations
We actively collaborate with experimental laboratories to validate computational predictions and drive new discoveries in molecular biology and biophysics.








Recent Publications
Dive into our blog for the latest trends, tips, and insights in the world of design and AI technology. Whether you’re looking for inspiration, tutorials, or industry news, our articles are crafted to keep you informed and inspired.


Funding
Our research is supported by grants from:
• National Institutes of Health (NIH)
• National Science Foundation (NSF)
Support our Research
Funding
Our research is supported by grants from:
• National Institutes of Health (NIH)
• National Science Foundation (NSF)
Support our Research
Funding
Our research is supported by grants from:
• National Institutes of Health (NIH)
• National Science Foundation (NSF)
Support our Research
Funding
Our research is supported by grants from:
• National Institutes of Health (NIH)
• National Science Foundation (NSF)
Support our Research